Simple protein-protein docking example
The simplest way to perform a protein-protein docking in LightDock is to use default parameters and to only provide two PDB files for both receptor and ligand. In this basic example, we will model the 2UUY complex starting from its unbound constituents.
IMPORTANT Please, make sure that you have the python3 version of LightDock installed (pip3 install lightdock). In order to perform your first protein-protein docking with LightDock, please follow the next steps!
Copying the data
Create a directory and copy the sample data provided.
cd ~/Desktop
mkdir test
cd test
curl -O https://raw.githubusercontent.com/lightdock/lightdock.github.io/master/tutorials/examples/2UUY/2UUY_rec.pdb
curl -O https://raw.githubusercontent.com/lightdock/lightdock.github.io/master/tutorials/examples/2UUY/2UUY_lig.pdb
LightDock setup
In previous versions of LightDock, a setup step was not required. From version 0.5.0 on, this is a mandatory step. To do so, please execute lightdock3_setup.py script to prepare your LightDock simulation.
lightdock3_setup.py
usage: lightdock_setup [-h] [--seed_points STARTING_POINTS_SEED]
[-ft ftdock_file] [--noxt] [-anm] [--seed_anm ANM_SEED]
[-anm_rec ANM_REC] [-anm_lig ANM_LIG] [-rst restraints]
[-membrane]
receptor_pdb_file ligand_pdb_file swarms glowworms
lightdock_setup: error: too few arguments
As you may notice from the displayed help, there are 4 arguments needed (the ones not enclosed by [ ]) to setup a quick LightDock simulation. These are:
- (1) receptor_pdb_file, the PDB file of your receptor molecule (usually the largest)
- (2) ligand_pdb_file, the PDB file of your ligand molecule (usually the smallest)
- (3) swarms, an integer corresponding to number of swarms to be generated
- (4) glowworms, an integer referring to the number of starting ligand conformations
For the sake of simplicity, we will generate 1 swarm containing 10 glowworms with the following command.
lightdock3_setup.py 2UUY_rec.pdb 2UUY_lig.pdb 1 10
[lightdock_setup] INFO: Reading structure from 2UUY_rec.pdb PDB file...
[lightdock_setup] INFO: 1628 atoms, 223 residues read.
[lightdock_setup] INFO: Reading structure from 2UUY_lig.pdb PDB file...
[lightdock_setup] INFO: 415 atoms, 55 residues read.
[lightdock_setup] INFO: Calculating reference points for receptor 2UUY_rec.pdb...
[lightdock_setup] INFO: Done.
[lightdock_setup] INFO: Calculating reference points for ligand 2UUY_lig.pdb...
[lightdock_setup] INFO: Done.
[lightdock_setup] INFO: Saving processed structure to PDB file...
[lightdock_setup] INFO: Done.
[lightdock_setup] INFO: Saving processed structure to PDB file...
[lightdock_setup] INFO: Done.
[lightdock_setup] INFO: Calculating starting positions...
[lightdock_setup] INFO: Generated 1 positions files
[lightdock_setup] INFO: Done.
[lightdock_setup] INFO: Preparing environment
[lightdock_setup] INFO: Done.
[lightdock_setup] INFO: LightDock setup OK
NOTE For a detailed description of the setup stage, please check the LightDock basics
If the setup is successful, we will find a file called setup.json, which includes all the details about LightDock simulation.
cat setup.json
{
"anm_lig": 10,
"anm_rec": 10,
"anm_seed": 324324,
"ftdock_file": null,
"glowworms": 10,
"ligand_pdb": "2UUY_lig.pdb",
"membrane": false,
"noh": false,
"noxt": false,
"receptor_pdb": "2UUY_rec.pdb",
"restraints": null,
"starting_points_seed": 324324,
"swarms": 1,
"use_anm": false,
"verbose_parser": false
}
This file is meant to be used for the simulation and reproducibility purposes. It can be also used to double check your simulation’s parameters prior running LightDock.
IMPORTANT You should not modify it unless you know what you are doing ;-)
Besides of setup.json, we find that several lightdock* files have been generated as well as an init/ and several swarm_* directories.
The init/ directory contains both the exact positions of the swarms (in this case a unique swarm: cluster_centers.pdb) and the starting positions of the glowworms (in this case 10 ligand conformations starting_positions_0.pdb). In the latter, the suffix 0 indicates the ID of the swarm.
Once the simulation is finished, the results will appear inside each of the swarm_* directories. Please refer to the following picture for a graphical description of the setup.
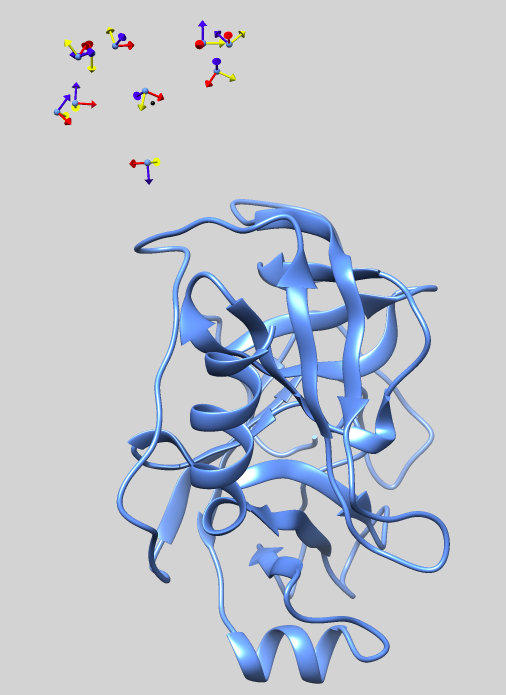
TIP If for any reason the setup stage fails, please remove all generated files before trying again. E.g: rm -rf lightdock* setup.json init/ swarm_*
LightDock simulation
Once the setup is successful, execute lightdock3.py script in order to run your first LightDock simulation. If you execute lightdock3.py without arguments a help menu will appear.
lightdock3.py
usage: lightdock [-h] [-f configuration_file] [-s SCORING_FUNCTION]
[-sg GSO_SEED] [-t TRANSLATION_STEP] [-r ROTATION_STEP] [-V]
[-c CORES] [--profile] [-mpi] [-ns NMODES_STEP] [-min]
[--listscoring]
setup_file steps
lightdock: error: too few arguments
There are 2 mandatory arguments (the ones not enclosed by [ ]) to run a quick LightDock simulation. These are:
- (1) setup.json, the file containing all parameters needed for the simulation
- (2) steps, an integer referring to the number of steps (
steps)
In this case, we will only perform 10 steps of GSO optimization with the following command.
lightdock3.py setup.json 10
@> ProDy is configured: verbosity='info'
[lightdock] INFO: simulation parameters saved to ./lightdock.info
[lightdock_setup] INFO: Reading structure from 2UUY_rec.pdb PDB file...
[lightdock_setup] INFO: 1628 atoms, 223 residues read.
[lightdock_setup] INFO: Reading structure from 2UUY_lig.pdb PDB file...
[lightdock_setup] INFO: 415 atoms, 55 residues read.
[lightdock] INFO: Loading scoring function...
[lightdock] INFO: Using DFIRE scoring function
[lightdock] INFO: Done.
[kraken] WARNING: Number of cores has not been specified or is incorrect. Using available cores.
[kraken] INFO: Kraken has 4 tentacles (cpu cores)
[kraken] INFO: Tentacle ready with 1 tasks
[kraken] INFO: Tentacle ready with 0 tasks
[kraken] INFO: Tentacle ready with 0 tasks
[kraken] INFO: Tentacle ready with 0 tasks
[kraken] INFO: 1 ships ready to be smashed
[lightdock] INFO: Monster spotted
[kraken] INFO: Release the Kraken!
[kraken] INFO: folding tentacle Tentacle-2
[0] step 1
[kraken] INFO: folding tentacle Tentacle-4
[kraken] INFO: folding tentacle Tentacle-3
[0] step 2
[0] step 3
[0] step 4
[0] step 5
[0] step 6
[0] step 7
[0] step 8
[0] step 9
[0] step 10
[kraken] INFO: folding tentacle Tentacle-1
[kraken] INFO: 1 ships destroyed
[lightdock] INFO: Finished.
By default, and if no other scoring function is specified, LightDock makes use of the DFIRE scoring function. However, we recommend to instead use its fast implementation fastdfire. For a complete list of the current supported scoring functions, please run lightdock --listscoring.
As you may have noticed, there is a warning on the number of CPU cores used [kraken] WARNING: Number of cores has not been specified or is incorrect. Using available cores. By default, LightDock will look for the total number of available cores. If you want to specify a different number, use the flag -c NUMBER_CORES. MPI is also supported using the -mpi flag
NOTE For more information about the simulation stage, please check the LightDock basics
LightDock results
If the run is successful and the [kraken] is back to sleep, we will find the output files for each of the independent swarms. In this case since we only generated 1 swarm, the results will be inside swarm_0. The output files will be named as gso_X.out, being X the step number. In our case, under swarm_0 we will only find 2 output files (gso_0.out and gso_10.out).
NOTE LightDock will only store the results every 10 simulation steps (0, 10, 20, 30, …)
In each of the output files, every line corresponds to a glowworm agent in the algorithm. The numbers enclosed by ( ), refer to the x,y,z coordinates in the translational space + the quaternion vector (q = a + 0i + 0j + 0k) in the rotational space. If ANM were enabled, this vector would expand by the number normal modes considered for receptor and ligand respectively.
The coordinates of the docked complexes are followed by the ID of the complex and the last column refers to the scoring, in this case as calculated with DFIRE.
head -2 swarm_0/gso_10.out
#Coordinates RecID LigID Luciferin Neighbor's number Vision Range Scoring
(31.4171143, 1.8570079, -6.3956223, -0.1058407, -0.4849369, 0.5997430, -0.6276482) 0 0 11.25395618 0 4.200 7.52800101
Generate docked models
Finally, to generate the resulting docked structures in PDB format, we will use the script lgd_generate_conformations.py. We will need to run this script for each of the generated swarms and provide two different arguments:
- (1) lightdock_output, the output file (usually the last one)
- (2) glowworms, an integer indicating the number of complexes to generate (=< number of glowworms used in the simulation)
Please note that, it is only possible to generate the docked conformations according to a single output file.
TIP If you want to generate the full trajectory for a given swarm, you should independently generate the conformations for every output file.
cd swarm_0
lgd_generate_conformations.py ../2UUY_rec.pdb ../2UUY_lig.pdb gso_10.out 10
@> ProDy is configured: verbosity='info'
[generate_conformations] INFO: Reading ../lightdock_2UUY_rec.pdb receptor PDB file...
[generate_conformations] INFO: 1628 atoms, 223 residues read.
[generate_conformations] INFO: Reading ../lightdock_2UUY_lig.pdb ligand PDB file...
[generate_conformations] INFO: 415 atoms, 55 residues read.
[generate_conformations] INFO: Read 10 coordinate lines
[generate_conformations] INFO: Generated 10 conformations
Inside the swarm_0 folder, we will find 10 new PDB structures corresponding to the 10 glowworm agents used in the current example.
CONGRATS! You have successfully run your first docking simulation with LightDock!
References
For a more complete description of the algorithm as well as different tutorials, please refer to LightDock, or check the following references:
-
Integrative Modeling of Membrane-associated Protein Assemblies
Jorge Roel-Touris, Brian Jiménez-García & Alexandre M.J.J. Bonvin
Nat Commun 11, 6210 (2020); doi: https://doi.org/10.1038/s41467-020-20076-5 -
LightDock goes information-driven
Jorge Roel-Touris, Alexandre M.J.J. Bonvin and Brian Jiménez-García
Bioinformatics, Volume 36, Issue 3, 1 February 2020, Pages 950–952, doi: https://doi.org/10.1093/bioinformatics/btz642 -
LightDock: a new multi-scale approach to protein–protein docking
Brian Jiménez-García, Jorge Roel-Touris, Miguel Romero-Durana, Miquel Vidal, Daniel Jiménez-González and Juan Fernández-Recio
Bioinformatics, Volume 34, Issue 1, 1 January 2018, Pages 49–55, doi: https://doi.org/10.1093/bioinformatics/btx555