The LightDock Server
LightDock is a fully open-source framework for flexible protein-protein, protein-peptide and protein-DNA docking, based on a swarm intelligence optimization algorithm: Glowworm Swarm Optimization (GSO). The LightDock Server is a fully rewritten version of the LightDock framework into the Rust programming language for optimal speed and performance. Here, we show the general features of the service and will provide an step-by-step guide on how to submit and analyze a docking run.
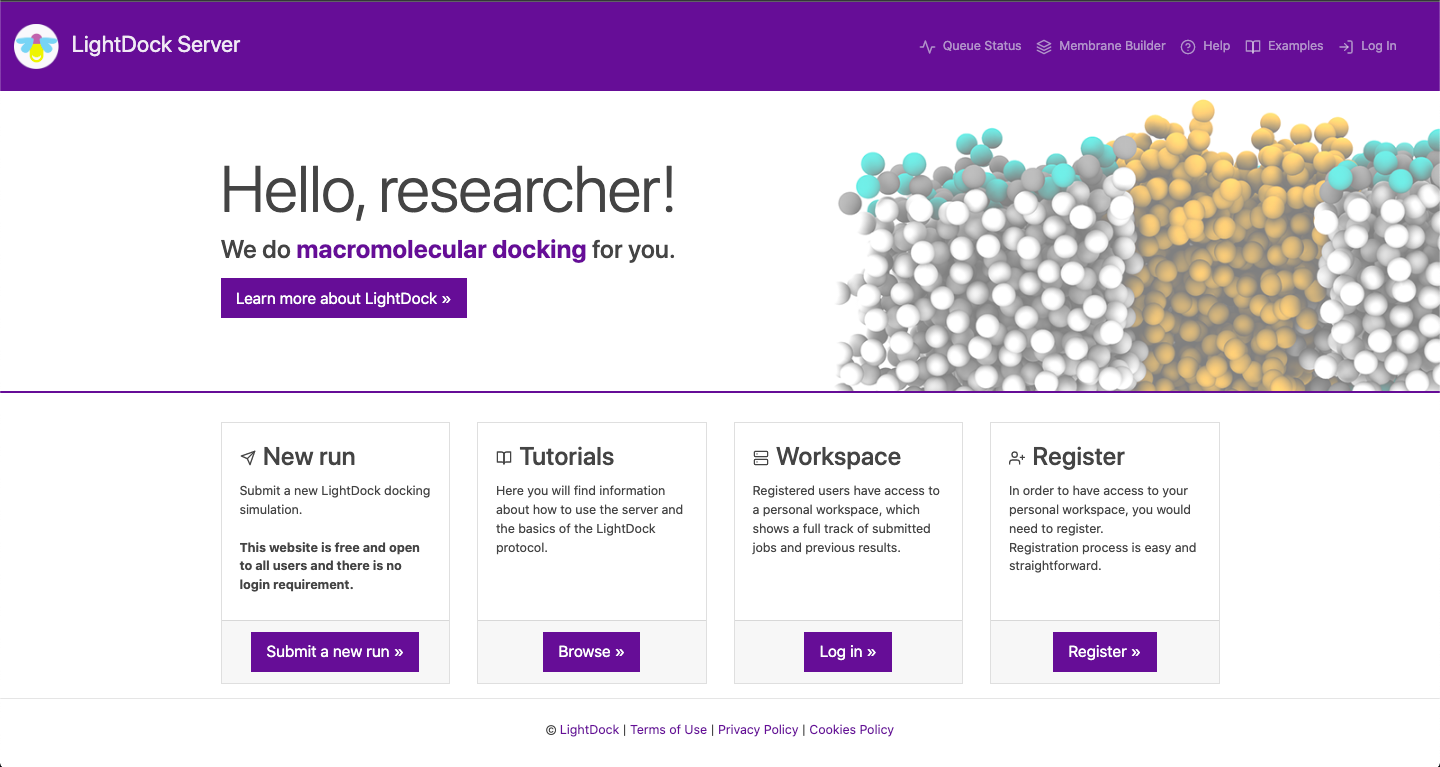
This is the landing page. In the top left corner you can find the LightDock logo that, when clicked, it will get you back to the landing page. In the top left corner you can check the status of the queue, make use of our in house membrane builder, get help about certain features of the server, check four different examples with pre-calculated data and log in into your personal workspace.
You can also learn more about LightDock; the docking engine underneath the server. Moreover, you might check an extensive list of tutorials for the standalone version of the software and register, in order to have access to your personal workspace. Again, registration is not required. Finally, you can submit a docking run by clicking on Submit a new run.
Step 1
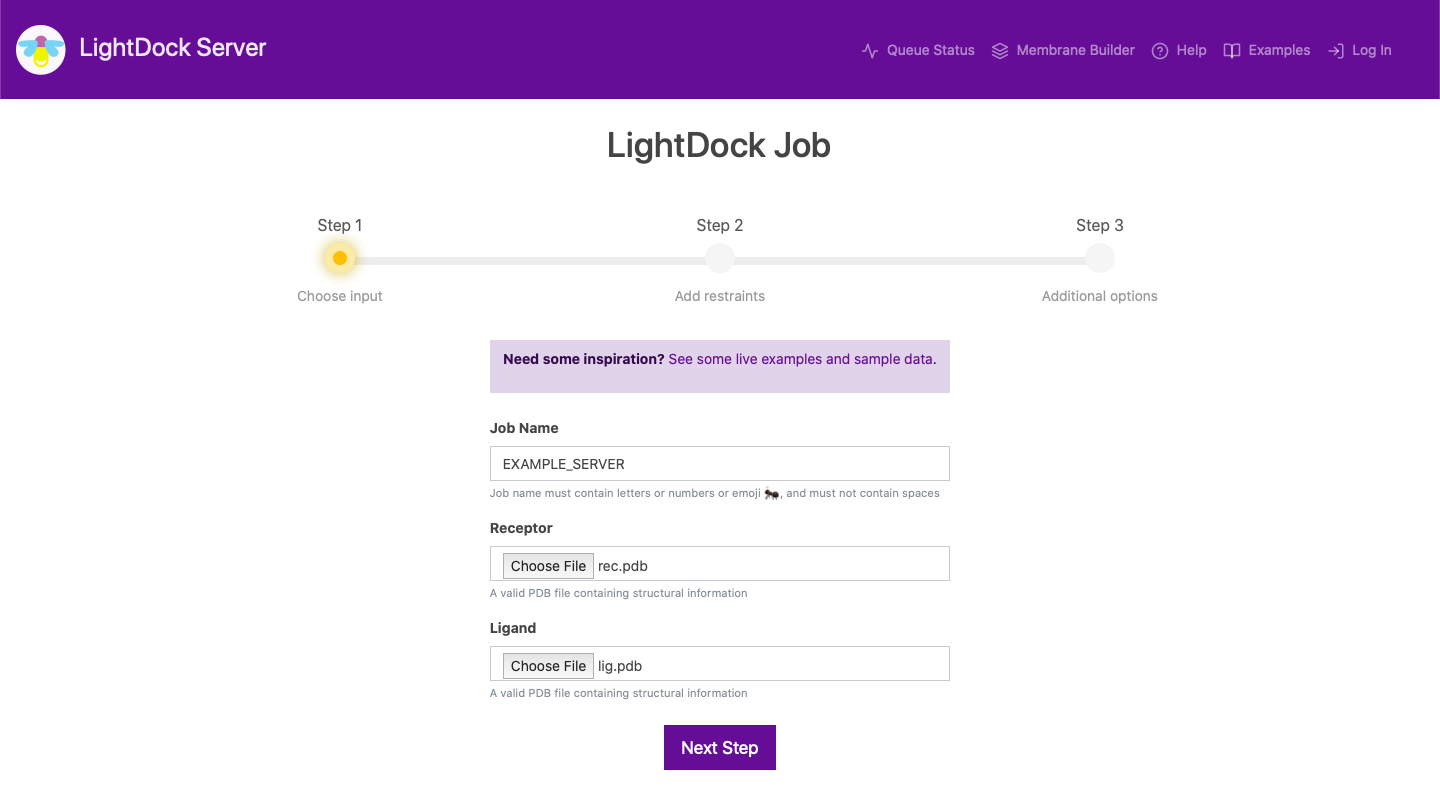
First, you are asked to input a job name and two PDB formatted files for your receptor and ligand molecules. These molecules can be proteins, peptides or nucleic acids. After clicking on Next Step, and if there are no inconsistencies on the data, you will be directed the next step.
Step 2
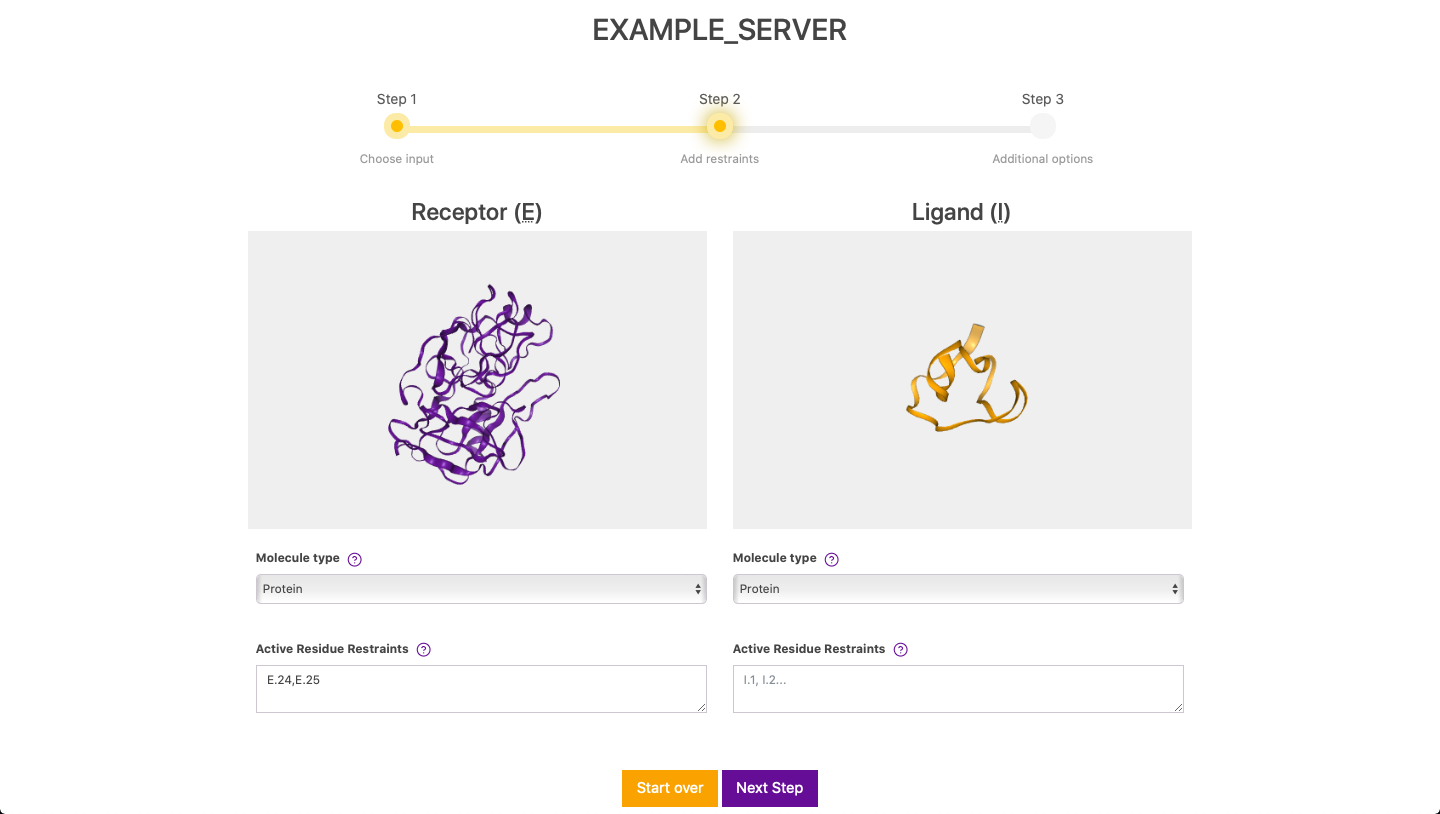
Here, you are able to inspect your molecules and you are required to select the molecule type of your molecules. In this case, we select Protein for both the receptor and ligand. Next, if wanted, you can define residue restraints for either the receptor or both the receptor and ligand. Retraints are defined in the form of E.24,E.25, where E stands for the chain ID and the number for the residue number. Please note that defining restraints only on the ligand is not allowed to avoid wasting precious computational resources. If this is the case, simply swap both inputs on Step 1 by clicking on Start over. After clicking on Next Step and if there are no inconsistencies on the data, you will be directed the next step.
Step 3
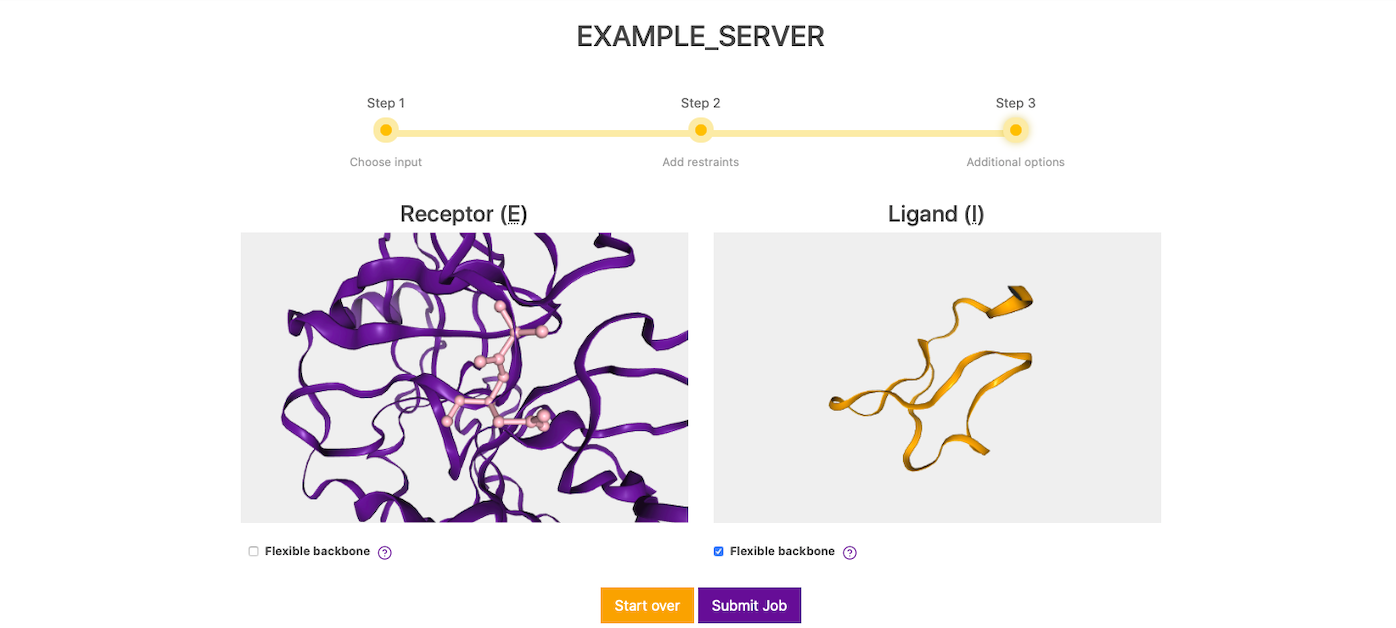
In this screen, the residue restraints defined in the previous step will show as ball and stick representation (pink) and molecules can be inspected for the last time before submission. Also, in this step you can enable (disabled by default) flexibility on either or both molecules. Once everything is set, you can submit your docking by clicking on Submit Job.
Results

Once submitted, the server will inform you about the status of your run. This is the time where you should bookmark the URL in order to retrieve the resultslater on. If you are a registered user, the job will be automatically stored in your personal workspace.
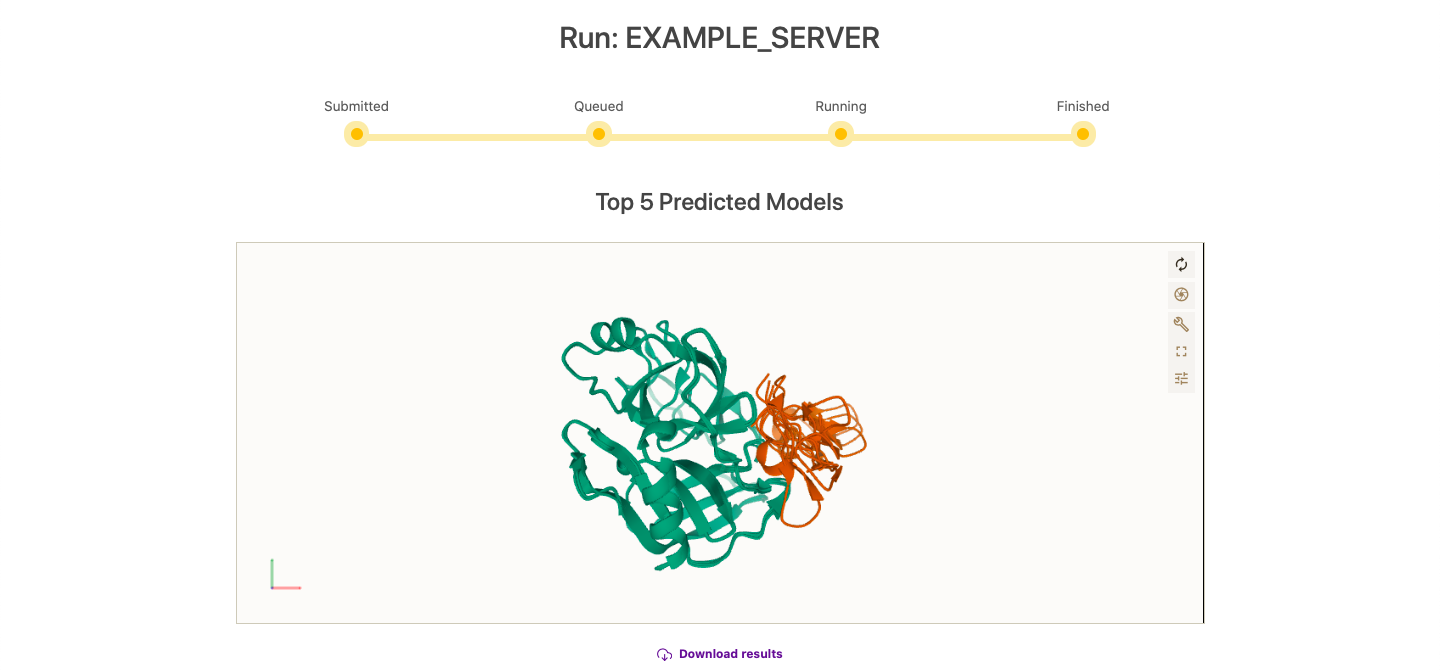
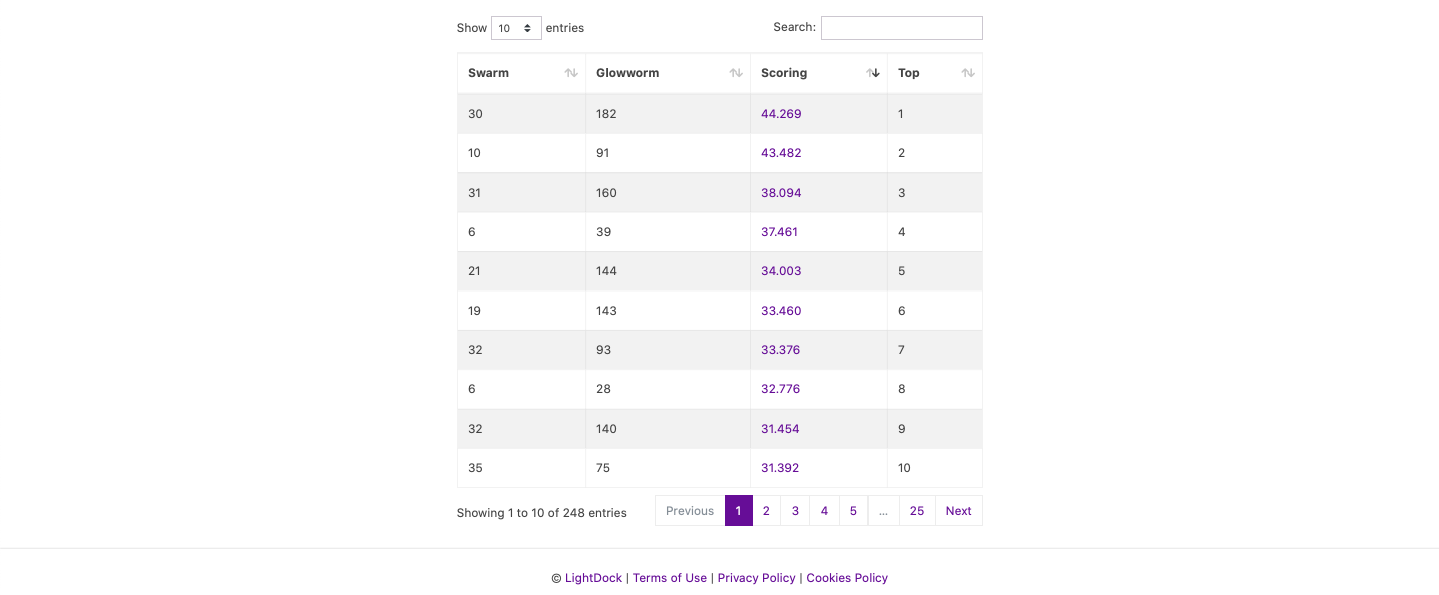
As soon as the run finishes, results will automatically show. The Top 5 models will show in the graphical interface for inspection. Moreover, the full run might be downloaded as a compressed file. On the bottom of the results page, all results are showed in a table and each individual complex can be downloaded by clicking on its corresponding scoring value.
Workspace
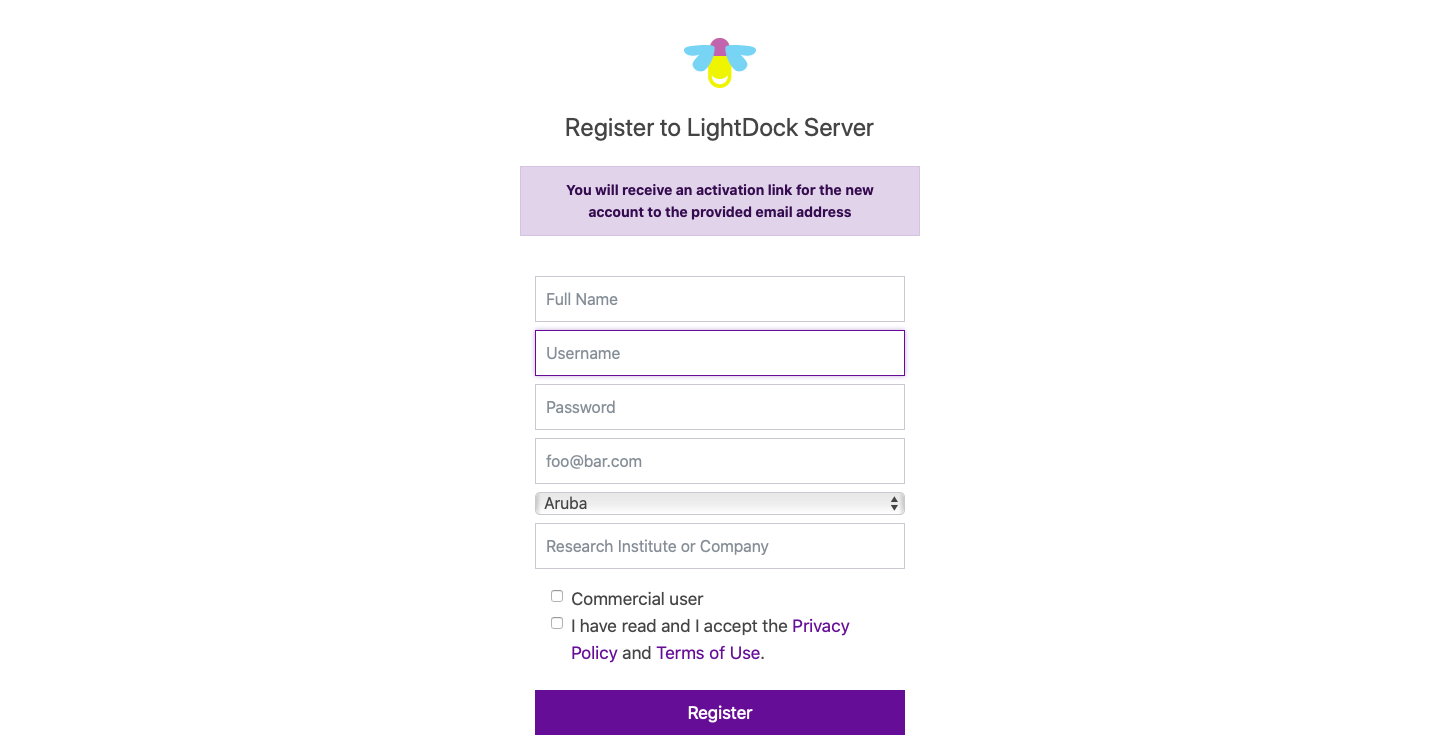
In order to get access to a personal workspace, you must first register. Please, check our privacy policy in advance. As shown in the screenshot above, you must indicate your full name, provide an username and password, as well as a valid email address together with your country of residence and research institute or company.
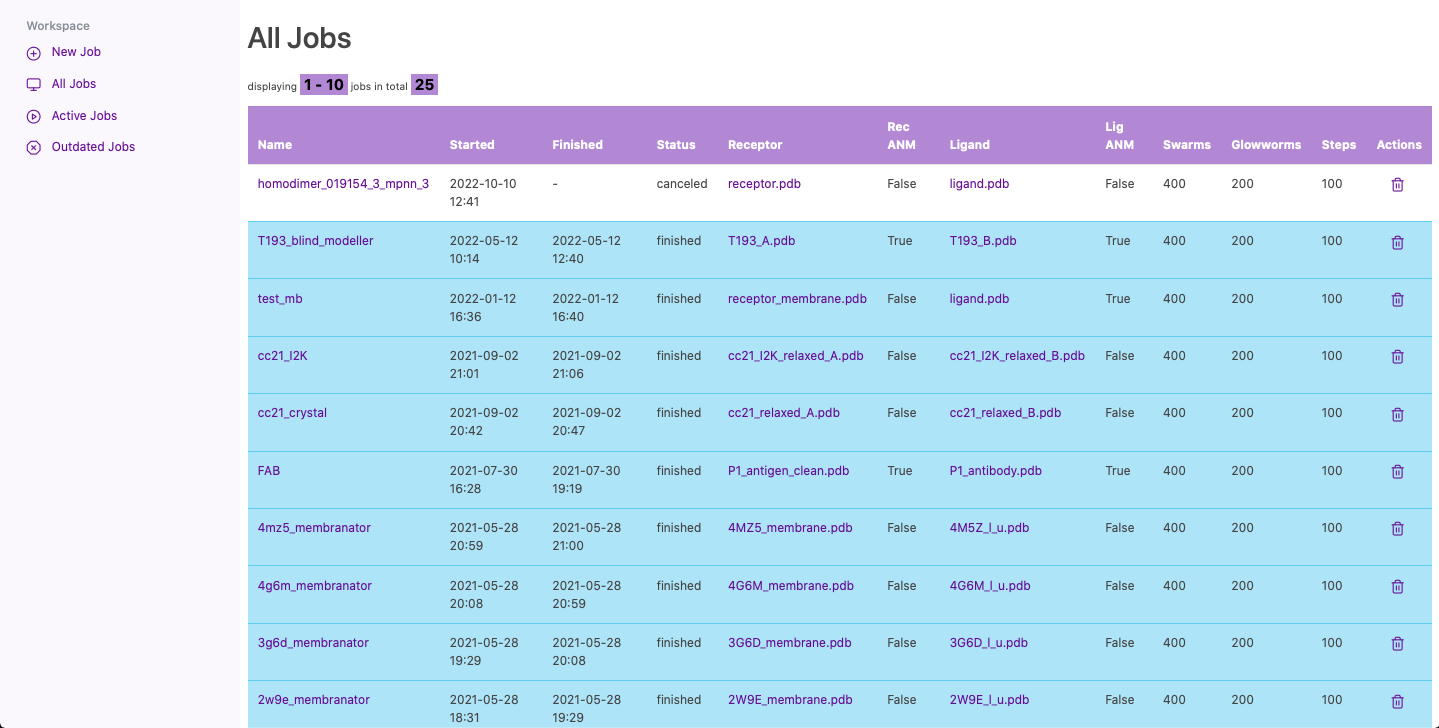
Once your account is activated, you can log in into your personal workspace. In this space, you will be able to manage past, present and future jobs. You can start a new job, or list all of your active or outdated jobs. Also, you can go to the results page by clicking on the job name or you can cancel/delete any of your jobs.
Should you have questions, please join us on Slack.