Docking Challenge 🏆

1. Introduction
Integrative structure modeling (ISM) computationally combines data from multiple sources of information with the aim of obtaining structural insights that are not revealed by any single approach alone (Braitbard, Schneidman-Duhovny & Kalisman, 2019).
With the aim of practicing and testing the ISM capabilities of the LightDock software, we propose the following challenge:
Are you ready to model the molecular completo mojado?
2. The challenge
We will model a molecular completo mojado by using a camelid nanobody-peptide complex as scaffold. The PDB of the complex is 6I2G.
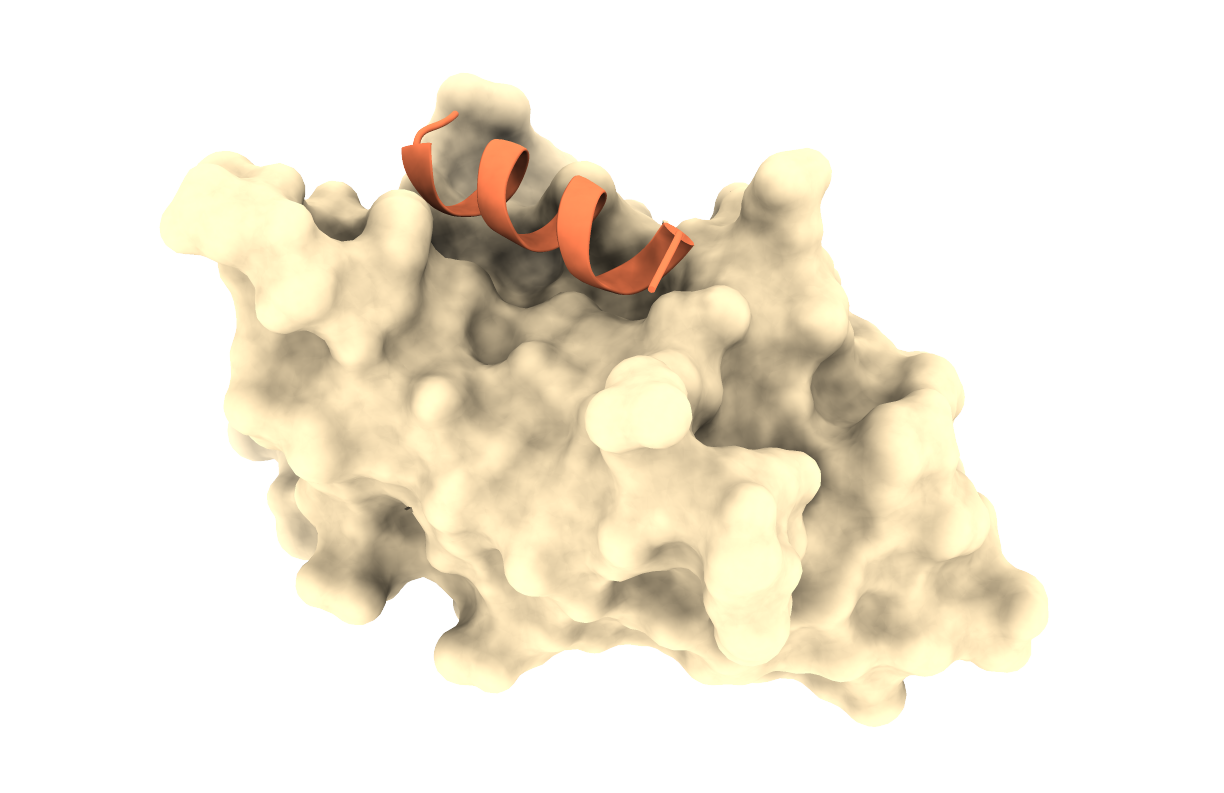
2.1. The bread: 🍞
We have prepared for you the scaffold receptor structure: 6i2g_nanobody.pdb

2.2. The stuffing: 🥩🥬🍅
You will need to model a T-A-L-C-A peptide:
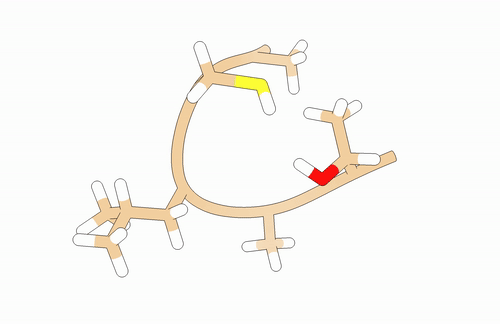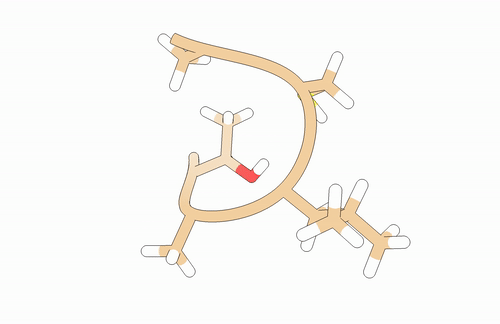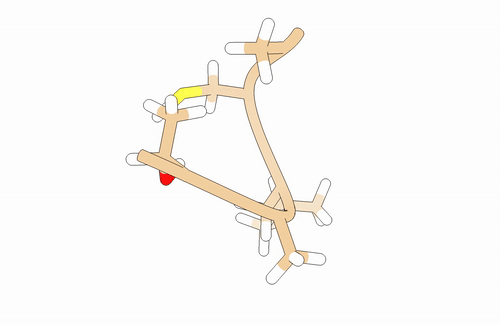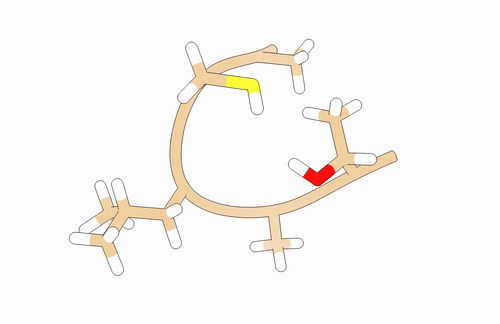
You may use pyPept Python package for building the peptide. Secondary structure is just a proposal!
If you don’t manage to build the peptide, you may download the one we have already prepared: talca_peptide.pdb
2.3. The goal: 🌭
Is that a completo? Is it mojado? Oh, wait, it is our completo molecular mojado!

3. Tips & tricks
1️⃣ Blind docking will probably help since we are able to recover a similar interface (see a simulation we have prepared for you).
2️⃣ Docking simulations tend to maximize/minimize scoring functions by increasing the number of interactions. This usually favors larger binding interfaces, it might be the case for this challenge.
3️⃣ Residue restraints might definitely help to place our ligand in the good binding interface, but this is a totally new interaction compared to the native complex.
We have prepared a notebook ready to run on Google Colab. Have fun!