Membrane-associated protein docking
- 1. Integrative modeling of claudin-19 in complex with the C-CPE enterotoxin
- 2. Protocol
- 3. Behind the scenes
- 4. Results
1. Integrative modeling of claudin-19 in complex with the C-CPE enterotoxin
Claudins are multipass membrane proteins with a major role in cell adhesion. The Clostridium perfringens enterotoxin (C-CPE) can bind to certain claudins, triggering tight junction disintegration and increasing permeability across epithelial cell sheets. It is reasonable to assume that the C-CPE enterotoxin will bind to the extracellular domain(s) of Claudin-19 to disrupt these cell–cell connections.
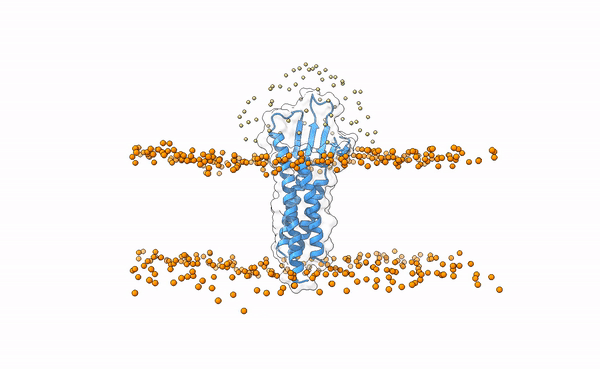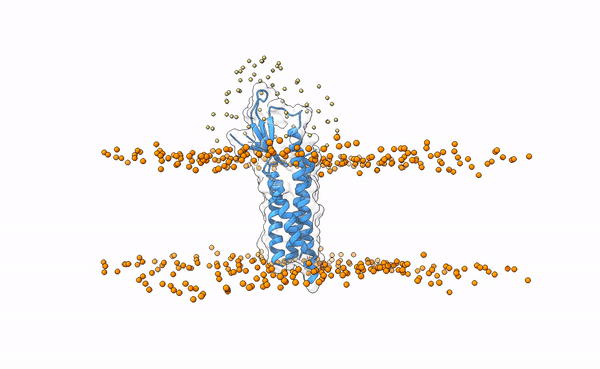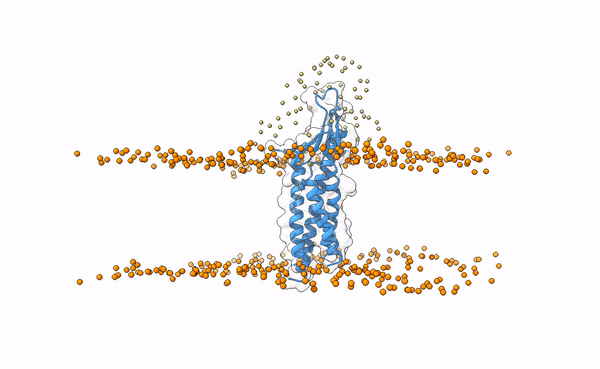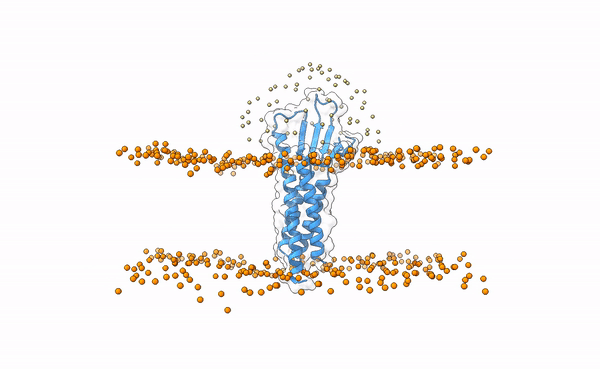
In this tutorial we will be working with the crystal structure of Mus musculus Claudin-19 transmembrane protein (PDB code 3X29, chain A) in complex with the unbound C-terminal fragment of the Clostridium perfringens Enteroxin (PDB code 2QUO, chain A). The PDB code of the complex is 3X29 (chains A and B).
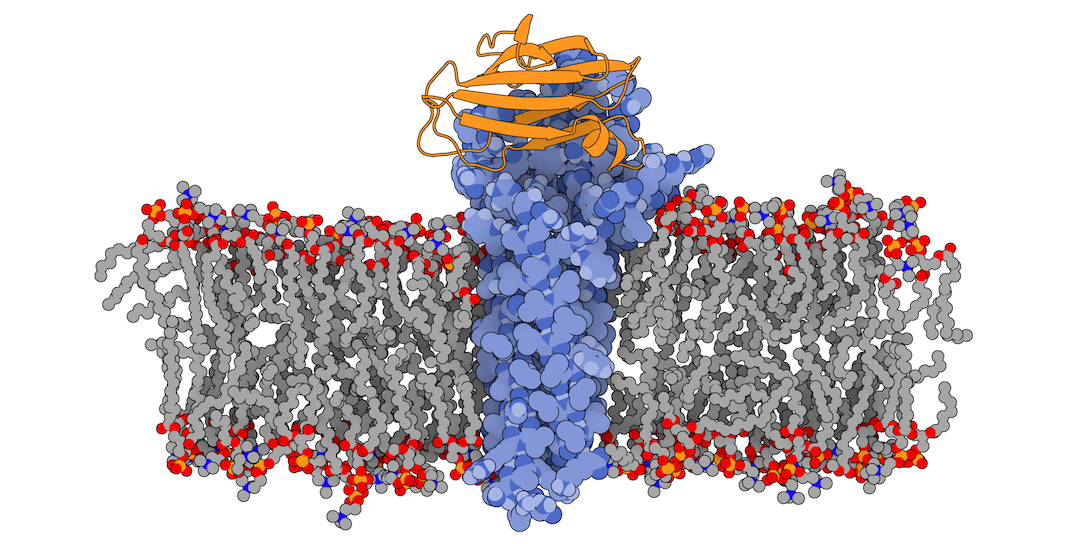
3X29 complex in a lipid bilayer as simulated by MemProtMD.
2. Protocol
Assuming that there is no information available in the MemProtMD database, we can instead use sequence-derived information from UniProt and our in-house Membrane Builder utility (aka Membranator) to build an estimated bead layer embedding Claudin-19.
The topological information from the UniProt entry indicates that Claudin-19 contains four helical transmembrane regions (segments 8–28, 82–102, 118–138 and 161–181) and five extramembranous regions: three cytoplasmic (segments 1–7, 103–117 and 182–224) and two extracellular (segments 29–81 and 139–160).
Download the PDB file corresponding to the Claudin-19 receptor. Go to Membranator and using the information retrieved from UniProt, select a residue as anchor for generating the membrane (e.g. S138) and default parameters. After inspecting the resulting model you may now download it. For your convenience, you can also download it from here
Having the receptor file ready, download the unbound C-terminal fragment of the Clostridium perfringens Enteroxin and proceed to the landing page of The LightDock Server. If you have an account, you may now log in. Alternatively, you can submit the run without the need of registering.
2.1. Step 1
Choose a name for your Run (e.g. protein-membrane) and upload both your receptor and ligand files and click on Next Step. Please, make sure that you are uploading the receptor that contains the estimated membrane generated by Membranator.
2.2. Step 2
At this point, you have the opportunity to visualize your molecules in an interactive mode. In this case, this is of great importance for the receptor as the positioning of the membrane will directly influence the generation of the swarms. Notice that for the receptor, the Molecule type: Membrane protein has been automatically assigned, as the server correctly detects the presence of a membrane. Similarly, for the ligand the Molecule type: Protein has been automatically assigned. In this case, there is no other possibility. For a complete list of use cases, check our publication. Here you also have the chance to specify residue restraints for both partners, but we will these boxes empty for this specific case. Click on Next Step.
2.3. Step 3
Finally, you may want to specify additional parameters, such as backbone flexibility. In this case, flexibility for the receptor is disabled by default as protein and membrane as treated as the same entity. Nervertheless, you can enable it for the ligand by clicking on Flexible backbone. After accepting the Privacy Policy and Terms of use, you are all set to submit your simulation by clicking on Submit Job.
3. Behind the scenes
While the simulation is running on the server, we take the opportunity to briefly explain how this protocol works.
The following image shows the distribution of the swarws around the theoretical binding region. This area has been entirely defined by the position of the membrane as we have not used any other kind of information (i.e. restraints). By doing this, we bias the sampling towards the relevant regions, and therefore discard some other areas where we know the interaction is not taking place. This speeds up the simulation while increases the chances of recapitulating the complex by reducing the amount of false positives in the final pool of docked poses.
Distribution of swarms in the current simulation.
Besides the initial swarms’ configuration, in LightDock the membrane also plays a crucial role during the optimization process. We use it internally as physical barrier, avoiding the ligand crossing it. We do this by applying a penalty to those models with overlapping (trespassing) residues, and thus biasing the optimization of them towards model with better scores (i.e. with hopefully not overlapping residues).
4. Results
While the simulation is done, we will have the opportunity to inspect the results on the server. If minimization has been selected, note that the membrane beads have been removed. Else, they will stay. At a first glance, we see that the simulation has converged towards a binding site, exploring somewhat different orientations of the ligand, and that the binding site is not overlapping with the position of the membrane. See picture below for details.

Here, you can check the results of the simulation.